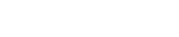與藥品研發(fā)(R&D)領(lǐng)域的科學創(chuàng)新相似,在過去的二十年中��,中國通過監(jiān)管創(chuàng)新對藥品法規(guī)進行了重大變革���,以加快藥品審評審批���。而中國創(chuàng)新藥物的蓬勃發(fā)展,尤其受益于支持創(chuàng)新的政策���。統(tǒng)計研究發(fā)現(xiàn)�,從2005年到2021年��,隨著具有新作用機制的藥物出現(xiàn)�����,包括免疫檢查點抑制劑和細胞治療產(chǎn)品�,進口和國產(chǎn)新型抗癌藥物的數(shù)量都顯著增加。
與2006-2010年相比��,2016-2020年進口藥品的藥品滯后期也大幅縮短了70%以上。最近���,來自于NPMA和清華大學等機構(gòu)的學者發(fā)文詳細分析了中國抗癌藥法規(guī)演變與監(jiān)管創(chuàng)新�,以及其對中國抗癌藥物和創(chuàng)新藥物開發(fā)的影響����。
NMPA由來
自成立以來,中國的藥品監(jiān)管機構(gòu)在過去的二十年中不斷發(fā)展�。1998年,國家藥品監(jiān)督管理局(SDA)成立�����,以解決跨省藥品審批標準不一致問題���,這標志著法律制度的開始(圖1)��。1999 年�����,SDA 首次發(fā)布并根據(jù)需要進行修訂的《中國臨床試驗質(zhì)量管理規(guī)范指南》確立了臨床研究的質(zhì)量和完整性標準�����。2003年��,食品行政管理職能并入國家食品藥品監(jiān)督管理局���,由該局轉(zhuǎn)變?yōu)閲沂称匪幤繁O(jiān)督管理局。隨后的國家食品藥品監(jiān)督管理總局(CFDA)成立于2013年��,升格為國務(wù)院部級機構(gòu)�����。
2018 年���,作為中國政府行政改革的一部分���,CFDA 成為新成立的國家市場監(jiān)督管理總局下屬的“國家藥品監(jiān)督管理局”(NMPA)。藥品審評中心(CDE)隸屬于國家藥監(jiān)局及其前身����,負責藥品(包括化學、生物制品和中藥)的臨床試驗申請��、上市許可申請以及后續(xù)修訂和續(xù)展的科學評估��。為了使其結(jié)構(gòu)更符合現(xiàn)代化醫(yī)藥,CDE實施了多輪機構(gòu)重組�,以簡化審查程序。到 2016年底����,CDE的員工數(shù)量翻了兩番,并且進一步擴大��,這也為更高效的審查流程做出了巨大貢獻�。通過采取一系列監(jiān)管創(chuàng)新來加速藥物開發(fā)和藥物審評,監(jiān)管機構(gòu)正在成為一個以科學為基礎(chǔ)���、以臨床價值為基礎(chǔ)的監(jiān)管機構(gòu)���,旨在確保藥物的有效性、安全性和質(zhì)量�����。鑒于與各種惡性腫瘤相關(guān)的未得到滿足的醫(yī)療需求不斷上升�����,抗癌藥物一直是藥物研發(fā) (R&D) 的重點�,它代表了大多數(shù)加快項目指定的藥物���,并在中國新批準的藥物中占據(jù)主導地位。
在這里����,來自于NMPA和清華大學的研究者統(tǒng)計分析了2005年至2021年中國抗癌藥物批準的趨勢和特征����,以說明監(jiān)管創(chuàng)新對藥物研發(fā)的影響。
圖1:Iteration of China’s key pharmaceutical laws and policies since 1998
關(guān)鍵法律和政策的迭代
中國的藥品監(jiān)管框架由多個層次組成��。規(guī)范藥品(包括生物制品)的主要法規(guī)是《中華人民共和國藥品管理法》(DAL)����,并輔以一套通用實施細則,簡稱《藥品管理法實施條例》����。DAL之下,藥物注冊條例 (DRR) 是管理臨床試驗和藥物注冊的核心文件����。另外, CDE 同樣參與起草與藥物開發(fā)和注冊相關(guān)的指導文件��。在過去的幾十年中,針對歷史事件�,立法和政策進行了重大的迭代和調(diào)整。監(jiān)管法規(guī)最初基于第一版《藥品管理法》(DAL�,1984 年頒布,2001 年修訂)和 DRR(2002年頒布�����,2005年生效)��。2007年��,在發(fā)生多起藥物事故后�����,如鞘內(nèi)注射被長春新堿污染的甲氨蝶呤引起的嚴重神經(jīng)系統(tǒng)不良反應(yīng)��,當局對 DRR 進行了實質(zhì)性修改�����,以加強對藥品質(zhì)量的監(jiān)管����。在接下來的十年中����,直到2015年都沒有對DAL和DRR進行進一步的全面修訂���。
2015年��,隨著公眾健康需求與審批步伐緩慢的矛盾不斷加劇,監(jiān)管改革的號角開始吹響�����。在標志性文件《關(guān)于改革藥品醫(yī)療器械審評審批制度的意見》(國發(fā)〔2015〕44號)和《關(guān)于深化審評審批制度改革鼓勵創(chuàng)新的意見》�����,及《藥品醫(yī)療器械管理辦法》(國發(fā)〔2017〕42 號)的指導下�,一系列前所未有的舉措出臺,用于提高藥品審評效率��、激發(fā)臨床價值創(chuàng)新�����、提升藥品質(zhì)量等��。為了與監(jiān)管創(chuàng)新的實施相結(jié)合,CFDA 于2017年作為監(jiān)管成員加入國際協(xié)調(diào)委員會 (ICH)����,并承諾在中國實施國際標準。在鼓勵創(chuàng)新藥的同時�����,監(jiān)管機構(gòu)也鼓勵仿制藥����,同時通過提高仿制藥質(zhì)量一致性評價要求規(guī)定的技術(shù)標準來強調(diào)藥品質(zhì)量。
為適應(yīng)藥品研發(fā)的快速發(fā)展��,國家藥監(jiān)局逐步采用以臨床價值為基礎(chǔ)的藥品監(jiān)管體系���。升級后的系統(tǒng)現(xiàn)在正在引領(lǐng)行業(yè)重視創(chuàng)新并關(guān)注患者的需求��。DAL和 DRR的最新修訂版(分別于 2019 年 12月1日和 2020年7月1日生效)納入并優(yōu)化了經(jīng)過時間考驗和有效的監(jiān)管流程�����,包括快速指定程序����、與監(jiān)管機構(gòu)的溝通機制以及 60個工作日的默許(IND)通過程。越來越多的科學和技術(shù)指南已經(jīng)發(fā)布��,以輔助新修訂的DAL和DRR���,而且在2021年��,CDE更是創(chuàng)紀錄的發(fā)布了87條指南����。值得注意的是����,最近頒布的一項指南強調(diào)了抗癌藥物研發(fā)中“臨床價值”的驅(qū)動力��,這為以患者為中心的藥物開發(fā)奠定了基礎(chǔ)���。此外���,中國國家衛(wèi)健委發(fā)布了關(guān)于藥物安全性、有效性���、經(jīng)濟價值���、創(chuàng)新性�����、適宜性和可及性的全面���、科學的藥物評價框架試點指南。
中國獲批的抗癌藥物
2005年1月至 2021年5月���,中國當局批準了660種抗癌藥物���,其中包括 94 種新藥(表 1)。2005-2007年期間���,絕大多數(shù)(241/258)為仿制藥�����,13種新藥上市����。2008年,受多起藥品事故和腐敗案件的觸發(fā)���,主管部門決定加強藥品管理和監(jiān)管��,對仿制藥實行集體審評��。2008 年的集體審查和 2016年的GQCE要求是顯著提高仿制藥標準和排除多余仿制藥的兩個最重要的里程碑�。結(jié)果�,多個臨床申請被申辦者自愿撤回。此外�,注冊積壓影響了新藥和仿制藥的批準。在2017年之前�����,每年批準的抗癌藥物數(shù)量顯著下降����。
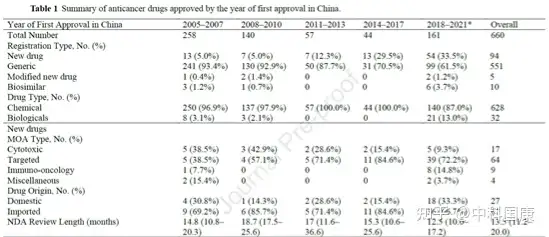
特別是在2014-2017年期間�,僅批準了13種新藥和31種仿制藥(表1)。隨著監(jiān)管改革和研發(fā)能力的提高���,中國自2018 年以來出現(xiàn)了藥物開發(fā)的爆炸式增長(圖2A和 B��,表1)�。值得注意的是,2018年至 2021 年5月�,獲批的新藥數(shù)量達到54個(33.5%)(表 1)。同期獲批仿制藥99個����,生物類似藥6個,改良新藥2個�。同樣,在研的候選藥物的數(shù)量在近幾年也顯著增加����。
作為藥物研發(fā)中的監(jiān)管限速步驟,IND和NDA審查時間大大縮短����。根據(jù) 2013年CDE 年度報告24,化學品和治療藥物IND申請的平均等待期分別為6個月和13個月�。自從實行60個工作日的默許審批制度以來,據(jù)報道�,2021年 99.86% 的IND申請在時限內(nèi)獲得批準。在新藥申請 (NDA) 審評時長方面���,抗癌新藥申請時長的中位數(shù)從2008-2010年的18.7(17.5-25.6)個月下降到 2018年1月-- 2021年5月的12.5(10.0-17.2����,P=0.0028)(下圖A)。2008-2011年提交IND申請的新藥獲得上市許可的總中位時間為 8.1(7.3-9.1)年�����,而2012–2015年提交IND申請的該時間減少到 5.3(4.8-5.9���,P<0.0001)年(下圖 C)�����。
不斷增長的科技創(chuàng)新促進了具有多種作用機制(MOA)的藥物的開發(fā)(表1)�����。2010年之前���,中國獲批的抗癌藥大多為細胞毒化療藥物,且以仿制藥居多���。在接下來的十年中,細胞毒藥物的數(shù)量有所下降�,而其他 MOA 的藥物數(shù)量卻在增加��。具體而言�����,新靶向藥物的數(shù)量一直在增長��,從 2005-2007 年的5個產(chǎn)品(38.5%)增加到 2014-2017 年的11個產(chǎn)品(84.6%)��,并在2018年至 2021 年5月期間達到 39個產(chǎn)品(72.2%)�。隨后 ����,靶向仿制藥和生物仿制藥最近顯示出快速增長,2018 年至 2021 年5月批準了40種仿制藥和6種生物仿制藥�。免疫療法在 2018 年開始蓬勃發(fā)展,截至 2021 年5月有8種產(chǎn)品獲批���。值得注意的是����,兩種嵌合抗原受體 (CAR) -T細胞療法于分別于2021年6月和2021年9月在中國獲批�����。
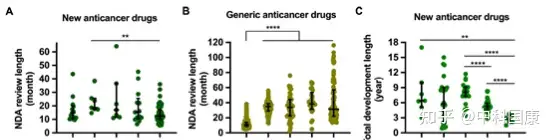
圖2:Trend of NDA review length and total development length of new anticancer drugs approved in China, 2005 to 2021
表1:Summary of anticancer drugs approved by the year of first approval in China.
推動國產(chǎn)藥物研發(fā)
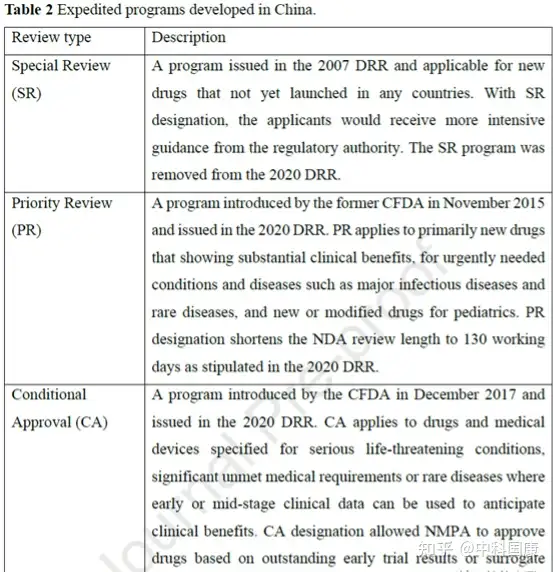
自2007年以來,中國制定了多個加速項目��,用以不斷促進國內(nèi)創(chuàng)新����。第一個行動是在行業(yè)和監(jiān)管機構(gòu)之間建立溝通渠道。特別審查 (SR) 計劃最初是在2007年修訂的DRR中引入的�,用于治療嚴重疾病并顯示出實質(zhì)性臨床益處的新藥(圖1,表2)���。具有SR稱號的藥物將在 CDE 的開發(fā)和注冊方面得到更嚴格的指導��。研究發(fā)現(xiàn)��,39種(41.5%)獲批的新抗癌藥物被授予SR稱號(圖 3)��。
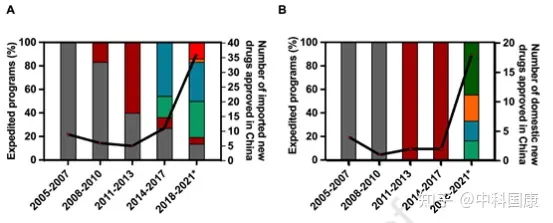
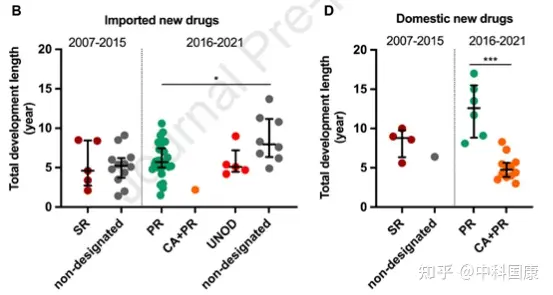
SR指定側(cè)重于溝通���,但對NDA審查長度和總開發(fā)長度幾乎沒有影響(圖4)。自 2016年發(fā)起人與監(jiān)管機構(gòu)之間建立正式溝通渠道后���,該計劃的好處變得不那么明顯��。因此�,SR指定在2020年終止��。2015年12月�����,作為全面監(jiān)管改革的先驅(qū)措施之一��,優(yōu)先審評(PR)項目��,可以有效的縮短藥品審評時間��。PR計劃的目標最初是為了減少注冊積壓�,隨后在 2017年12月轉(zhuǎn)向提高臨床價值,可以解決未滿足的醫(yī)療需求并顯示出大量臨床益處的藥物有資格獲得這一稱號����。2017年后批準的18個(100%)國產(chǎn)抗癌新藥獲得PR(圖3),中位審評時間為14.6個月(圖4)�。
圖3:Uptake of expedited programs (EP)
圖4:NDA review length and total development length of approved new anticancer drugs in China by expedited programs, 2007 to May 2021
監(jiān)管機構(gòu)引入了兩個額外的計劃來增加監(jiān)管靈活性,包括有條件批準 (CA) 和突破性治療 (BT)(表 2)�。2017 年12月,基于可合理預測臨床益處或不太全面的臨床數(shù)據(jù)的替代終點建立了用于批準藥物的CA途徑���。CA適用于在危及生命或罕見疾病的情況下��,臨床價值高于現(xiàn)有療法的藥物�����。例如����,與第一代抑制劑依魯替尼相比,可能具有療效和安全性優(yōu)勢的兩種國產(chǎn)新一代布魯頓酪氨酸激酶 (BTK) 抑制劑 zanubrutinib 和 orelabrutinib 獲得 NMPA 有條件批準用于復發(fā)/難治性慢性淋巴細胞白血病患者或小淋巴細胞性淋巴瘤����。
與美國的加速批準計劃類似,在中國接受CA的藥物需要在上市后繼續(xù)確認其實際臨床益處�����。統(tǒng)計結(jié)構(gòu)表明���,CA 指定與減少的總開發(fā)時間長度顯著相關(guān)(圖 4D)�����。12種國內(nèi)PR指定藥物在CA途徑下獲得批準���,IND 提交后的中位數(shù)為 4.8 (3.9-5.7) 年���,而國內(nèi)PR指定藥物未接受CA的時間為 12.6 (8.9-15.5) 年(差異,7.8 年; P = 0.0002)����。BT是在2020 年修訂的DRR中首次引入的稱號����,該稱號給予初步臨床數(shù)據(jù)顯示比現(xiàn)有療法有很大的臨床益處的藥物。BT 指定藥物的開發(fā)可以通過與當局的密切溝通來加快開發(fā)進程�����,這些藥物可能有資格獲得CA和PR途徑����。截至 2022年5月,46種抗癌新藥獲得中國BT認定��。其中5款已獲 NMPA 批準用于 相關(guān)適應(yīng)癥����。
表2:Expedited programs development in china
減少藥物滯后
促進進口新藥的可及性是解決未滿足的醫(yī)療需求的另一種方式。在 2000年之前,當確定持久的醫(yī)療需求時�����,無需在中國進行額外的臨床試驗即可批準藥物�����。隨著研發(fā)和監(jiān)管經(jīng)驗的積累��,當局開始要求在上市批準之前提供中國數(shù)據(jù)�����。2017年之前�,對于新藥而言,在海外開展2期試驗是中國機構(gòu)參與多中心臨床試驗的前提��,在中國上市前必須持有其他國家頒發(fā)的上市許可證�����?����?鐕扑幑疽虼送ㄟ^橋接研究將全球批準的藥物引入中國,該政策在2008年至2013年期間主導了中國的注冊策略�����。
此外��,低效的藥物審評過程是進口藥物進入的另一個障礙�����。調(diào)查結(jié)果表明�,2008-2013年進口新藥的NDA審評時間最長���,為17.7(15.9-22.6)個月��。正如 2013 年CDE年度報告所報告的那樣�����,NDA審查的積壓情況越來越多(長達14 個月的等待期)����,而進行橋接研究的化學藥物IND審查的等待期甚至更長(長達 19 個月)�。因此���,中國的藥物研發(fā)和批準顯著落后于西方,2001-2005年在美國開始臨床試驗的抗癌藥物在中國滯后5.8(3.4-7.8)年進入臨床階段���;) 2006 -2010年�����,在美國批準的抗癌藥物進入中國滯后6.6(2.8-9.5)年�����。這種藥物滯后(定義為一種新藥在美國獲批和隨后在中國獲批之間的時間)限制了藥物的可及性并阻礙了藥物的創(chuàng)新��。
圖5:Clinical development and first approval of imported new anticancer drugs inthe US and China
自2015年以來���,中國監(jiān)管部門采取了多項開創(chuàng)性措施來改善中國的監(jiān)管和研發(fā)環(huán)境(圖 1)。并且自2017年10月起�,監(jiān)管結(jié)構(gòu)允許在中國參與者中對在中國境外開發(fā)的新藥物進行同步的首次人體研究(圖 1)。此外�����,2017年加入ICH為中國注冊技術(shù)要求與全球標準的統(tǒng)一鋪平了道路���。這些措施有利于中國早期參與多中心臨床試驗��,促進患者可及性����。隨后,采用 MRCT 途徑的藥物比例開始擴大��,在 2015年至2021年5月期間達到52%��。與橋接臨床實驗相比��,采用MRCT的藥物可以縮短中美之間藥物滯后的問題(3.8 VS 4.7 年��,P = 0.225)����。此外�����,中國一直在簡化審批程序��, PR指定新藥的審評時間大大縮短�����。對于 31/44 指定為 PR 的進口藥物,NDA審查時間中位數(shù)為12.0(10.2-15.5)個月���,而非指定藥物為19.4(11.8-25.9�,P = 0.040)個月(圖 4A)����。這也伴隨著相對較短的總開發(fā)時長(PR,5.7 [5.0-7.5] 年與非指定PR的8.0 [6.4-11.2] 年�����;圖 4B)�。
2018年國家藥監(jiān)局發(fā)布的指南正式采納了境外試驗臨床數(shù)據(jù)。2018年至2020年發(fā)布的加快批準外海外急需藥品(UNOD)清單就是一個典型的例證���。UNOD 指定的藥物通常具有顯著的臨床益處��,并且如果沒有種族差異�,則可以在有限甚至沒有中國臨床數(shù)據(jù)的情況下申請上市許可���,這可以有效縮短 NDA/生物許可申請 (BLA) 審查時間三到六個月�。UNOD 名單上的五種抗癌藥物在IND提交后上市的中位時間為 5.1(4.5-7.2)年(圖 4B),中位審查時間為 6.9(5.6-8.8)個月(圖 4A)���。其中�,2018年獲美國食品藥品監(jiān)督管理局(FDA)批準的FLT3抑制劑gilteritinib���,與挽救性化療相比�����,顯著提高了FLT3突變的復發(fā)或難治性(r/r)急性髓細胞白血病患者的生存率����。鑒于中國這些患者的治療選擇有限���,gilteritinib 于2020年11月被列入UNOD清單�����,并于2021年2月獲得NMPA批準。
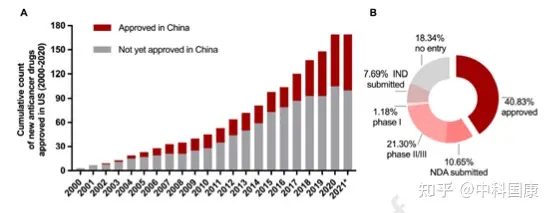
由于制藥行業(yè)����、臨床和監(jiān)管機構(gòu)的共同努力�,近年來藥物滯后時間明顯縮短�。伴隨著藥物批準差距的縮短(1.9 [1.0-2.9] 年,2016-2020年 vs. 6.6 [2.8-9.5] 年����,2006-2010年;P = 0.0006�,圖5),2011-2015年在美國進入臨床階段的抗癌藥物進入臨床階段的滯后時間急劇下降至 2.0 (1.4-3.5)年(相比之下�,2001-2005 年為5.8 [3.4-7.8]年,P = 0.0009 )����。擬合的 LOWESS 曲線顯示了藥物滯后減少的趨勢,到2021年接近1.4年(圖6)����。因此,在過去的二十年中����,美國獲得批準的新型抗癌藥物在中國獲得許可的比例一直在上升(圖7A)。截至2021年5月�,2000年至2020年間在美國獲得許可的抗癌藥物中,有40.8%(69/169)已在中國獲得上市許可,另有10.7%正在等待提交的 NDA/BLA審查(圖 7B)��。
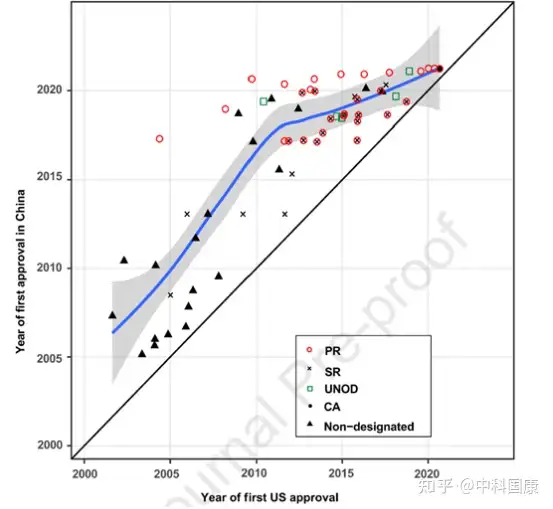
圖6:First approval dates of imported new anticancer drugs in the US and China
圖7:Current status in China of new anticancer drugs approved in the US
討論
經(jīng)過二十多年的改革����,中國的藥品監(jiān)管機構(gòu)已經(jīng)升級為一個更加高效、科學和以臨床價值為基礎(chǔ)的體系���,以跟上快速發(fā)展的科技創(chuàng)新步伐��。我們見證了越來越多的進口和國產(chǎn)抗癌新藥在中國獲批��,這在很大程度上得益于近期的監(jiān)管變革���、研發(fā)能力和技術(shù)創(chuàng)新。改善藥物可及性以解決未滿足的醫(yī)療需求一直是中國監(jiān)管機構(gòu)和藥物開發(fā)商的首要任務(wù)���。減少進口新藥的滯后是關(guān)鍵一步�����。如圖所示���,與美國相比�����,藥品滯后明顯減少,盡管進口藥品仍存在兩年滯后�。
改善藥物可及性以解決未滿足的醫(yī)療需求一直是中國監(jiān)管機構(gòu)和藥物開發(fā)商的首要任務(wù)。減少進口新藥的滯后是關(guān)鍵一步��。如上文所述���,盡管中國的進口藥品與美國相比仍存在兩年的進口滯后���,但與美國相比,藥品滯后顯著減少�。2020年最新修訂的DRR規(guī)定的并行藥物審查和基于風險的現(xiàn)場檢查預計將進一步縮短審查時間。Pralsetinib是平行審查的一個例子��,在PR和CA指定下審查長度為6.6個月��。此外��,向國內(nèi)企業(yè)授權(quán)藥品已成為海外生物科技企業(yè)搶先進入中國市場的有效途徑����。例如,Tesaro于2016年授予再鼎的niraparib 在 FDA 首次批準后的2.8年獲得 NMPA批準。Axicabtagene ciloleucel是一種CD19導向的CAR-T細胞療法���,是2021年6月在中國獲批的首個CAR-T細胞療法����,其技術(shù)是從Kite Pharma轉(zhuǎn)移過來的YESCARTA?(axicabtagene ciloleucel)�����。
近年來�����,本土創(chuàng)新蓬勃發(fā)展�����,但多數(shù)是一些 “me too”類藥物���。事實上�,適度的競爭對于衛(wèi)生系統(tǒng)控制成本和提高藥品負擔能力是必要的����,并且對于管理出現(xiàn)的藥品短缺也至關(guān)重要�。另外�����,患者也可以從一系列價格合理的抗癌藥物中受益����,包括me too��、仿制藥和生物類似藥��。然而�,類似藥物的重復開發(fā)會導致資源分配不當和對缺乏足夠藥物的治療領(lǐng)域的投資不足。例如����,中國已批準十余種抗 PD-1 或 PD-L1 單克隆抗體,但仍有大量類似的候選藥物處于臨床階段���。在仿制藥中已經(jīng)觀察到相似之處�����,如中國市場上 60 多種阿奇霉素片的仿制藥版本�,而即使在參考藥物的專利到期后,也沒有仿制藥硫酸阿扎那韋可用��。藥物開發(fā)的不平衡一直是一個全球性問題�。為了應(yīng)對這些障礙,F(xiàn)DA提供了一攬子監(jiān)管激勵措施�,例如指定孤兒藥,并建立了藥物短缺數(shù)據(jù)庫以監(jiān)測藥物可及性���。這些集體努力使美國在急需領(lǐng)域獲得批準的藥物越來越多��,包括數(shù)百個孤兒藥和涉及先前短缺藥物的仿制藥批準�����。同樣���,中國當局發(fā)布了監(jiān)管指南,強調(diào)未滿足的醫(yī)療需求��,并公布了幾份專利過期或接近專利到期且沒有仿制藥的藥品清單����。未來,在采購�、醫(yī)院使用和定價方面�,需要一個全面的價值和質(zhì)量評估框架�����,這將鼓勵開發(fā)具有實際臨床價值和高質(zhì)量仿制藥的創(chuàng)新藥����。
中國制藥行業(yè)目前正處于從“me-too”到“me-better”藥物的過渡期���?����;谒幬锝Y(jié)構(gòu)優(yōu)化或基于先進技術(shù)平臺的新藥將表現(xiàn)出更好的療效或安全性��,或為患者提供替代益處���。例如,世界上第一個皮下給藥的 PD-L1 抗體 envafolimab 可能比靜脈給藥的 PD-(L)1 抗體顯示出依從性和安全性優(yōu)勢�。
事實上,一流的創(chuàng)新不可避免地會給發(fā)起人和監(jiān)管機構(gòu)帶來更大的不可預見的風險��。適當?shù)娘L險管理和可靠的收益風險評估對于主動應(yīng)對這些挑戰(zhàn)至關(guān)重要��。令人鼓舞的是,中國的監(jiān)管體系不斷適應(yīng)外部變化����,做出科學的監(jiān)管決策。例如���,康方生物自主研發(fā)的“全球首創(chuàng)”雙特異性PD-1/CTLA-4抗體--卡多尼單抗��,基于其良好的初步數(shù)據(jù)獲得中國CDE的BT認定��,與來自其他PD-1+CTLA-4 抗體聯(lián)合療法數(shù)據(jù)相比���,在晚期宮頸癌的具有更高的客觀緩解率?����;谶@些優(yōu)異的結(jié)果��,該藥物已于 2022年6月通過加速途徑獲得批準����。這反映了中國公司在開發(fā) FIC 藥物方面取得的進展,以及審評人員在對具有新作用機制的藥物進行利益風險評估方面取得的進展���。
全球化正在成為中國藥物開發(fā)的常態(tài)�,除了將中國納入其全球注冊戰(zhàn)略的海外制藥公司,本土公司也開始走向國際舞臺����。2020年,在中國開發(fā)的抗癌產(chǎn)品中約有五分之一也在境外開發(fā)����。成功的全球發(fā)展需要嚴謹而前瞻性的戰(zhàn)略規(guī)劃,使用僅限中國的數(shù)據(jù)來支持其他國家的藥物營銷不可避免地具有挑戰(zhàn)性��。例如����,F(xiàn)DA 已經(jīng)對信迪利單抗和索凡替尼發(fā)出了完整的回復信�����。在做出監(jiān)管決定時��,F(xiàn)DA 曾表示��,未滿足的醫(yī)療需求程度和研究藥物的臨床獲益程度將決定監(jiān)管的靈活性程度��,這與中國的監(jiān)管機構(gòu)的觀點一致。值得注意的是����,zanubrutinib 為第一個實現(xiàn)跨國同步開發(fā)的國內(nèi)創(chuàng)新藥物,獲得了FDA和NMPA 的雙重批準��,并且其使用的關(guān)鍵數(shù)據(jù)主要來自中國患者���。第一個本土開發(fā)的ADC藥物disitamab vedotin 獲得FDA的BT指定��,用于HER2陽性局部晚期或轉(zhuǎn)移性尿路上皮癌的二線治療��。作為該適應(yīng)癥的一種新穎且有前景的治療方法�,disitamab vedotin 滿足了美國未滿足的醫(yī)療需求�。中國的NMPA一直在推動基于臨床價值的藥物開發(fā),要求使用可用的最佳護理標準作為對照����,并鼓勵及時就關(guān)鍵試驗設(shè)計和重大決策進行溝通。NMPA還致力于改進良好生產(chǎn)規(guī)范��,并于去年被接受為藥品檢驗合作計劃的預加入成員���。未來���,同步藥物開發(fā)和聯(lián)合監(jiān)管審查將進一步加速藥物研發(fā)的全球化����。
在整個藥物開發(fā)過程中���,全球多個監(jiān)管和衛(wèi)生社區(qū)一直在積極與患者互動�����。在美國�����,F(xiàn)DA 的以患者為中心的藥物開發(fā) (PFDD) 計劃被納入2012年重新授權(quán)的處方藥用戶費用法案 (PDUFA V) 和 2016頒布的21世紀治愈法案。FDA在實施有關(guān)患者參與的基本目標方面取得了重大進展�����,包括將患者體驗數(shù)據(jù)納入審查過程并提供PFDD指南���。建立了“Project Patient Voice”等新型網(wǎng)絡(luò)平臺�,以自愿報告患者的癥狀和副作用,補充臨床試驗數(shù)據(jù)的安全信息�����。在中國��,以患者為中心將成為常態(tài)���,近期腫瘤學研發(fā)指南中關(guān)于設(shè)計適合患者需求的臨床試驗和關(guān)于患者參與藥物研發(fā)的指南都強調(diào)了這一點��。此外���,中國還一直在采用分散臨床試驗(DCT)的方法來減輕試驗參與的負擔,使試驗?zāi)軌蜻B續(xù)進行����,監(jiān)管文件不斷形成,并對DCT要素提供更明確的指導����。
本站聲明:如果您認為轉(zhuǎn)載內(nèi)容侵犯了您的權(quán)益,請您來電聲明�����,我們將會在收到信息核實后24小時內(nèi)刪除相關(guān)內(nèi)容。